To: Main Page ; To: Application

To: Main Page ; To: Application

Go to evaluation of the output if you need a quick help to optimally run MotifSampler. |
Introduction |
|
MotifSampler is a probabilistic de novo motif detection tool designed to search for regulatory motifs in DNA sequences upstream of coregulated genes from one species. Compared to the first release of MotifSampler (3.1.1, Thijs et al. 2002), the 3.1.5 release of MotifSampler addresses some functional bugs, offers a wider range of prior probability distributions for estimating the number of instances per sequence (parameter -p) and improves the way the information of the internally computed distribution on the number of instances in a sequence is summarized in the algorithm.
The latest release (motifsuite_v2) can integrate position-specific prior (PSP) information to guide the motif search towards DNA regions that are more likely involved in active regulation.
|
Theoretical background of the algorithm |
|
MotifSampler belongs to the class of probabilistic motif detection programs that typically model motifs by a PWM (Position Weight Matrix) and the remainder of the sequence by a background model. The goal of such programs is to find the motif model that differs most significantly from the background model, therefore being a candidate functional (regulatory) motif in the input sequences dataset. 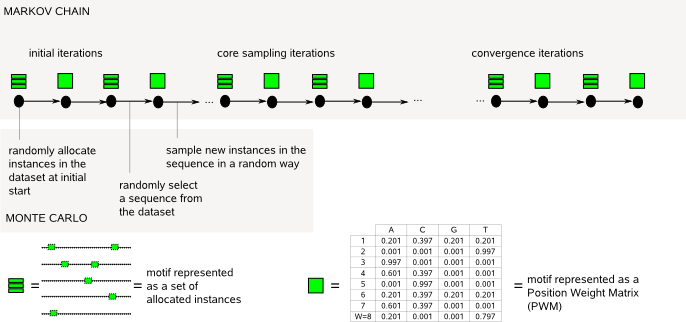 The central idea of how this is achieved is that the more accurate the description of one variable in one step, the more accurate will be the description of the other variable in the next step. So once some true instances have been selected by chance, the PWM starts to reflect the true motif and this process tends to recruit further true instances. The selection of instances is done by random sampling from a score distribution in such way that multiple combinations of different potential instances are sufficiently explored at the same time reducing the risk to quickly converge to a suboptimal solution. Because of the random property, the reported solution by MotifSampler may be (slightly) different if you repeat the iteration process on the same input sequence set with the same MotifSampler parameter settings. A true motif, however, will have a strong signal compared to the background and will be detected multiple times when repeating MotifSampler multiple times, as can be assessed with the post processing programs (MotifRanking, FuzzyClustering) available in MotifSuite. |
Input for the algorithm |
- a file (parameter -f) containing a set of DNA sequences in FASTA format. MotifSampler is designed to work on a set of DNA sequences that are expected to have a set of short sequences that are more overrepresented compared to the background in which they are hidden. Such sequences are typically derived from gene expression or ChIP-based experiments. |
MotifSampler Algorithm |
|
The algorithm of MotifSampler searches for one motif at a time. In its search for one motif, the algorithm is initialized by choosing exactly one instance of length w (parameter -w) at random in each sequence. It then proceeds through a sequence of (1) initialization iterations, (2) core sampling iterations to end with (3) convergence iterations (Fig.1). Each iteration in principle repeats the following two steps for every sequence in the dataset (Fig.2) : 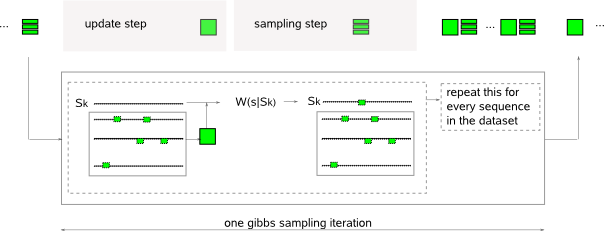 - update step : a set of instances is known at the end of the previous step. One sequence is excluded from the dataset (Sk) and a PWM is constructed based on this reduced set of instances. The reduced-PWM construction uses the nucleotide counts for A,C,G,T on each position in the reduced set of instances and a set of pseudocounts. The pseudocounts reflect a positional prior belief on A,C,G,T
(derived from the PSP information if so provided in inputfile -q)
and at the same time avoid the occurrence of singularities. 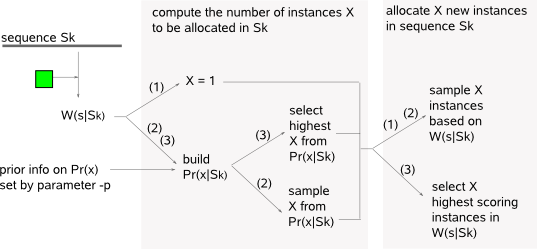 The random aspect of sampling in (1) and (2) enables the motif detector to better explore multiple possible solutions. High scoring instances have a higher probability to be sampled than low scoring instances and the closer we get to the best possible optimal solution in the dataset, the more informative will be W(s|Sk) and Pr(x|Sk) and the less likely the motif detector will iterate away from this solution. In the final phase (3), we assume that the motif detector is already near a good (hopefully the best) optimal solution in the dataset and by selecting the highest scoring values, the motif detector is guided towards the nearest optimal solution to end the detection process. The PWM and set of instances retrieved at the end of the convergence phase are the representations of 1 single candidate motif that are reported to the output files of parameter -m respectively parameter -o. 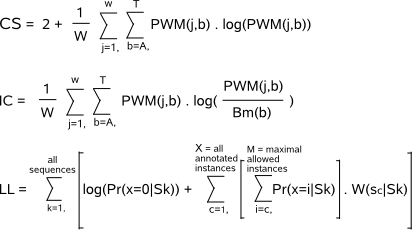 -CS is a measure for the conservation of the motif and this score can be used in absolute terms. A perfectly conserved motif has a score equal to 2 while a motif with a non-informative uniform nucleotide distribution has a score 0. |
Output to the user |
The algorithm reports two files that each describe the same set of candidate motifs detected by the algorithm in a different format : |
Evaluation of the output |
The number of solutions reported by MotifSampler is expected to equal the product of parameter -r and parameter -n. If there are less solutions reported, then MotifSampler did not converge to a reasonable solution (i.e. when the number of instances at the end of an iteration step is not higher than 2, the detection procedure is aborted). If the number of solutions obtained at MotifSamplers default settings is still sufficiently high to perform a proper statistical assessment (minimal 50 or 70 solutions on the safe side), we recommend to first run MotifRanking on the matrix output file (-m) to quickly see how frequently a motif was detected. If this frequency (return ratio RR) is sufficiently high (minimal 10% or 30% on the safe side), the retrieved motif can be regarded as a statistically significant solution that most likely represents a true motif. If no interesting motif was obtained, you should repeat MotifSampler with revised parameter settings (discussed below) that influence the way the motif detector walks and converges towards the most optimal solution in a dataset. |
Feedback |
|
Contact us if you have comments, questions or suggestions or simply want to react on the contents of this guideline. Thank you. |