To: Main Page ; To: MotifSampler, PHMS/NOMS

To: Main Page ; To: MotifSampler, PHMS/NOMS

Introduction |
Introduction |
|
While the present-day computational motif detection algorithms can accurately predict in vitro binding, solutions represent the in vivo reality more accurately when used in concert with additional regulatory knowledge. Integrating additional knowledge into the Gibbs sampling scheme can be challenging with computational or analytical complexities and uncontrolled outcome. Position-specific prior (PSP) information expresses a prior belief over DNA locations on how likely these positions are involved in active regulation. Using a PSP in the Bayesian framework of Gibbs sampling to guide the detection towards prioritized DNA regions is fast, easy and efficient. |
Integrate prior information in computational motif detection |
|
Active binding of a TF with its target TFBSs to perform a regulatory action is determined by a long list of characteristics that go beyond the sequence specificity of the potential binding (see Fig.1). For many of regulatory experiments, for example nucleosome positioning, ChIP-chip or ChIP-Seq peaks or DNaseI hypersensitive sites, the regulatory evidence can be converted into a prior probability over the location of potential motif sites. 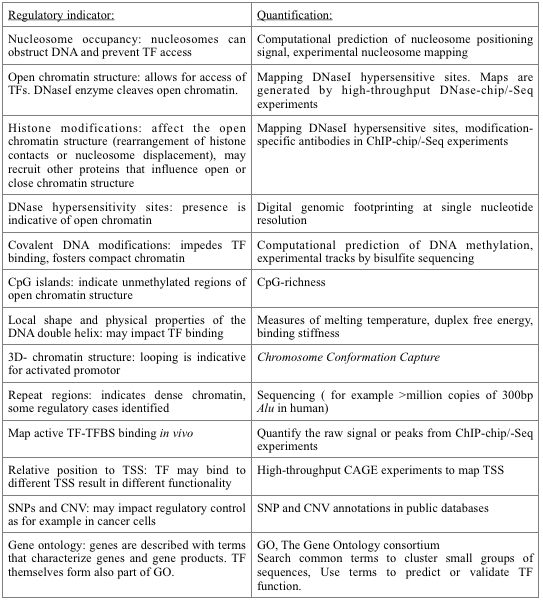 |
Impact of a PSP on the motif detection algorithm |
For reasons of simplicity, we describe further how we integrated the use of a PSP in MotifSampler. The same principles have been applied for the integration of a PSP in PHMS and NOMS. |
Using a PSP |
|
By its definition, a PSP is a sequence of numerical non-negative values that quantifies a distinct prior regulatory belief over DNA positions. The format of the file is defined in PSP file format. |
Special case: Create a conservation-based PSP |
|
Feedback |
|
Contact us if you have comments, questions or suggestions or simply want to react on the contents of this guideline. Thank you. |
References |
- Gordan, R., Narlikar, L., & Hartemink, A. (2009). Finding regulatory DNA motifs using alignment -free evolutionary conservation information. Nucleic Acids Research, 38(6):e90. |