To: Main Page ; To: Application

To: Main Page ; To: Application

|
Page contents : |
Introduction |
CreateBackgroundModel is designed to create a genome-specific higher-order background model for a set of non-coding DNA sequences. A genome-specific background model represents the probability of the nucleotides A,C,G,T to be non-functional non-coding data, also called background data, for the genome of interest. Building and using the right background model has a major impact on the performance of MotifSampler and MotifLocator. The CreateBackgroundModel method is independent of the application in which it is used afterwards so CreateBackgroundModel can be used on any list of sequences of which you want to model the nucleotide distribution. |
CreateBackgroundModel Algorithm |
Supply sequences (parameter -f) : 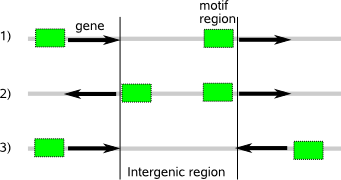 You can design such a sequences dataset for your organism of interest yourself by carefully selecting the non-coding region between two consecutive genes (intergenic region) available in published databases. The intergenic region can however be long and vary in composition for different organisms so you need to evaluate which subregion is most suitable. The direction of transcription of the two consecutive genes is also important (fig.1). When both genes are transcribed in the same direction, the intergenic region may contain motif instances that control transcription of the second gene in row. When the transcription direction is pointed in opposite directions away from each other, the intergenic region most likely has motif instances controlling the transcription of both genes. If the opposite transription directions point to each other, the motif instances are most likely outside the intergenic region. The latter sequences should be excluded from the analysis as they may not correctly represent the non-coding nucleotide distribution of the background where the motif signal is hidden in. Finally, the total length of all sequences should be significantly higher than the expected number and length of hidden motif instances so the construction of the background model is not biased towards the motif signal (making it more difficult for MotifSampler or MotifLocator to distinguish that motif signal from the background information where it is submerged in). 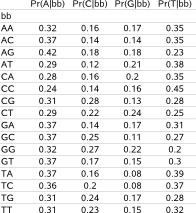
A background model of order 2 for example (fig.2), will have as first entry on the first row Pr(A|AA), the probability of having nucleotide A given the two preceding nucleotides are AA in the sequence, followed by Pr(C|AA), Pr(G|AA) and Pr(T|AA). The second row describes Pr(A|AC), Pr(C|AC), Pr(G|AC), Pr(T|AC) and so on untill the 16th row describing Pr(A|TT), Pr(C|TT), Pr(G|TT), Pr(T|TT). |
Output to the user |
|
The background model is written into a file in the following format : read format details. |
Evaluation of the output |
|
At all terms, the values in #oligo should all be sufficiently higher than zero as they represent the counts of preceding oligonucleotides that are necessary to compute the transition matrix. A subset of significantly low values in #oligo means that your input sequences dataset is not sufficiently long to correctly model the varying composition of the genome-specific background. The background model will be over-ordered and biased towards the input sequences dataset. This will result in a decrease of the (motif) signal to (background) noise ratio during motif detection. In those cases we recommend to repeat CreateBackgroundModel with a lower value for the order setting. So the higher the order of the background model, the more important it is to select the right set of input sequences for constructing the background model. |
Feedback |
|
Contact us if you have comments, questions or suggestions or simply want to react on the contents of this guideline. Thank you. |