To: Main Page

To: Main Page

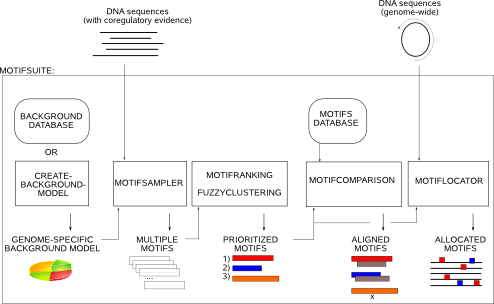
Step 1 : Fundamental input: promotor sequences and genome-specific background model |
|
As coregulated genes share some similarities in their regulatory mechanism, their promoter regions might contain some common motif instances that are binding sites for transcription factors (TF). A sensible approach to detect these regulatory elements is to search for overrepresented instances in the promoter regions of such a set of coregulated genes. Succesfull motif detection requires that the overrepresentation signal of the set of motif instances in the sequence set is sufficiently strong compared to the non-functional surrounding nucleotides, also called the background. A strong signal-to-noise ratio means that you optimally supply only those sequences where you have sufficient coregulatory evidence coming from e.g. cDNA microarrays , ChIP-Chip or ChIP-seq. 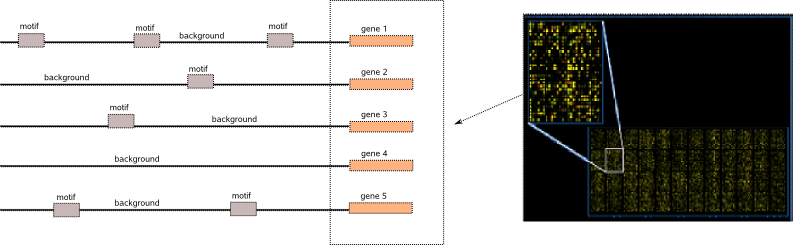
Probabilistic motif detection tools describe the sequence set using a motif model and a background model. The motif model is a position weight matrix (PWM) reflecting the probability to observe either one of the nucleotides A,C,G and T on each position of the overrepresented motif. The background model represents the probability of the nucleotides to belong to the remainder of the sequence set, assumed to be non-functional background data. It is the task of the motif detection tool to find the motif model that differs most significantly from the background model, therefore being a putative functional motif in the input sequence set. |
Step 2 : Probabilistic de novo motif detection: MotifSampler |
|
MotifSampler is a probabilistic motif detection tool that searches in the space of all possible overrepresented short sequences in the input sequence set (the search space) for the most overrepresented (most frequently conserved) motif. The search space may however contain several local optima, only some of which represent a true motif. Reducing the search space by setting the program parameters in a biologically relevant way will decrease the program runtime and will help guiding the algorithm towards those local optima that most likely correspond to true motifs. Important program parameters to set are for example the motif width, the number of different motifs and information (e.g. prior distribution and maximum) on the number of instances of each motif to search for per sequence. 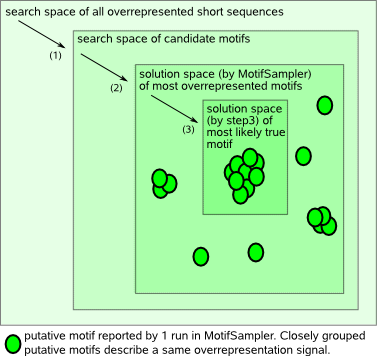 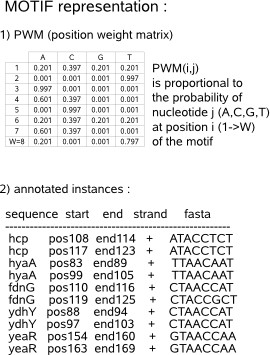 |
Step 3 : Summarizing motif detection results: MotifRanking and FuzzyClustering |
|
MotifRanking and FuzzyClustering are two complementary methods to summarize a list of multiple motif detection solutions reported for a given sequence set. Both methods follow the principle that only motifs that are found consistently in several motif detection runs are statistically pronounced and will most likely approximate a true motif. 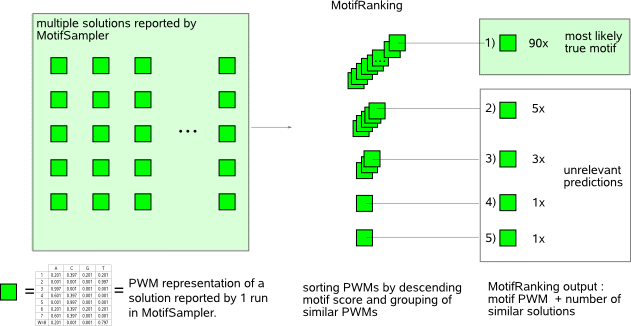 MotifRanking sorts the solutions (motifs) reported by MotifSampler in descending order of their motif score (default the LogLikelihood score computed by MotifSampler) and solutions that represent the same motif are grouped based on a matrix (PWM) comparison strategy. Grouping allows identifying the number of different motifs and counting how many times each of these respective motifs was detected by MotifSampler (thus assessing their significance). The motif with the highest motif score has the highest probability to be a functional regulatory motif provided it was detected with a minimal frequency amongst the multiple solutions reported by MotifSampler. 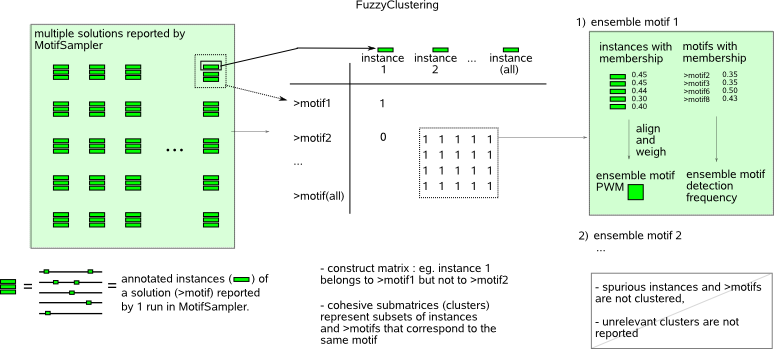 The multiple detected motifs and their instances listed in the input file are represented in a matrix (the matrix entries describe which instances belong to which detected motifs). FuzzyClustering uses a (spectral graph based) clustering technique to iteratively extract cohesive clusters (submatrices consisting of mainly non-zero values) from this matrix. Each extracted cluster stands for a set of instances and a set of detected motifs that all correspond to the same motif (ensemble motif). Each instance and each motif in the cluster get assigned a membership score that reflects how well the instance (motif) represents the ensemble motif. This method allows filtering instances (motifs) from the solutions reported by MotifSampler for which there is not enough evidence that they correspond to an overrepresented motif. |
Step 4 : Comparing motif models: MotifComparison |
You have now detected an overrepresented motif in a given sequence set, represented by a PWM (the instances format of the motif is not used in this step). MotifComparison will answer if your motif (also called the query motif) corresponds to any of previously described motifs reported in curated databases or to a motif detected by yourself in previous analyses.
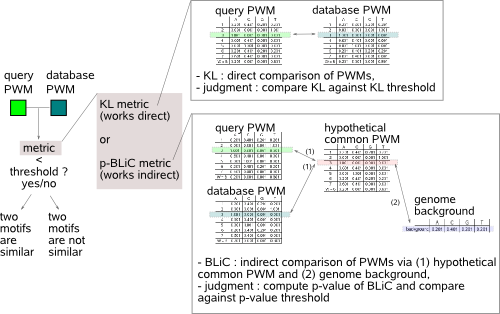
To do this, MotifComparison computes and judges on a similarity metric between the PWMs of the motifs to be compared. A first metric uses the well known and widely used Kullback-Leiber distance (KL, based on the mutual information between the PWMs that are being compared) and sets a threshold on KL to decide upon similarity. A second metric calculates a more advanced similarity score BLiC (Bayesian Likelihood 2-Component, Habib et al. 2008) that will not only compare the PWMs of the two motifs with a hypothetical common motif, but also with the background distribution of the genome where the motif applies to. In the BLiC score, motif positions where the nucleotide distribution (as described in the PWM) is similar to the background distribution are considered less relevant for motif similarity as they do not contribute to the sequence-specific binding of the motif. In addition, there must be significant evidence that a high BLiC score indeed means that the two motifs are similar. This is done by computing and setting a threshold on the p-value of the respective BLiC score, computed against a distribution of BLiC scores of randomly obtained non-similar motifs. |
Step 5 : Genome-wide screening with a motif model: MotifLocator |
|
In order to avoid too many false positive motif predictions, it is best to tune the parameters for de novo motif detection tools towards a regime that favors a high PPV (i.e. minimizing the prediction of false positive motif instances) at the expense of a lower sensitivity (i.e. missing true motif instances). To compensate for this low motif prediction sensitivity, you can use MotifLocator in a final step to screen your sequence set for missed instances. 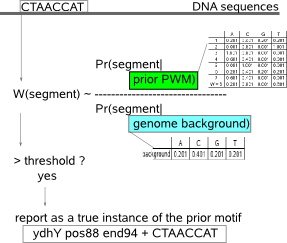 MotifLocator calculates for each possible instance in the sequence set how well it fits a given motif model (PWM) versus the genome-specific background model. A threshold on the background corrected PWM based score computed for each instance determines whether an instance in the sequence set is a motif or not. |
Feedback |
|
Contact us if you have comments, questions or suggestions or simply want to react on the contents of this guideline. Thank you. |