To: Main Page ; To: Application

To: Main Page ; To: Application

Go to evaluation of the output if you need a quick help to optimally run MotifComparison. |
Introduction |
MotifComparison is designed to compute the similarity between two motifs coming from two input files (a query file and a database file).
MotifComparison can be used on any two lists of motifs as long as the motifs to be compared are in the PWM format
used throughout MotifSuite. |
MotifComparison Algorithm |
The inputs for MotifComparison are two files each containing a (list of) motif(s) represented by a Position Weight Matrix (PWM).
A PWM is a matrix describing the probability -preferably described by a frequency (value between 0 and 1)
but also counts (numbers higher than 1 and also zeros are common) are allowed- to find any of the nucleotides A,C,G,T on each position of the motif
(PWM format describes conversion requirements in case your input file was generated outside MotifSuite). In what follows
we call the 'unknown' motifs the query motifs which are listed in the query file (parameter -m). The database file (parameter -d) lists
the motifs coming from a public or personal database, further called database motifs. When you automatically use precompiled database files,
you must verify that both files use the same type of PWM entries (or frequencies, or counts). 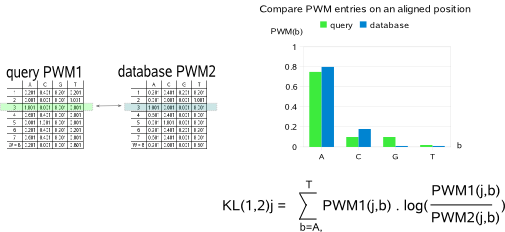 Each aligned position individually contributes to the KL distance between query and database motif. To compensate for differences in length of the query and database motif, the sum of the KL distances computed at each aligned position is averaged over the length of the overlapping part between the two aligned motifs. As different alignments between two motifs may give a different KL distance, all possible alignments that respect the minimum required overlap (parameter -x) and the maximal allowed shift (parameter -s) are evaluated (Fig.2). 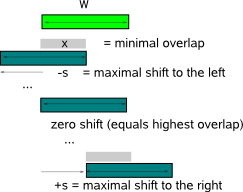
In default program mode, -s allows an alignment shift of only 1 nucleotide
and -x requires an alignment overlap of at least 6 nucleotides. The reason why two parameters -s and -x are introduced is mainly to handle motif
comparisons of different motif length : allowing for sufficient shifts while conserving a minimal overlap between aligned regions accomodates
the comparison of longer motifs (for which many shifts might be needed to find the optimal alignment) while the minimal overlap is needed to
constrain the allowed shifts for shorter motifs. When all input motifs have the same length, only one parameter is needed to
restrict the admitted alignments: the minimum overlap constraint (-w) is typically set to half of the (fixed) motif length and the maximal shift (-s)
to an unrestricting high value (e.g. equal to the motif length). Mark that multiple alignments also include alignments of the query motif with
the reverse complement of the database motif, as the query motif may also be transcribed in the opposite direction as described in the input file. 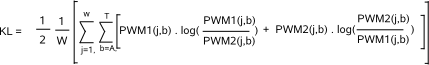 The more the PWMs of the query and database motif are similar, the lower will be the KL distance between these two motifs. The KL distance will be
zero if the two motifs being compared have perfectly equal PWMs. Finally, two motifs are judged similar if the KL distance between their PWMs is
below a given similarity threshold (parameter -t). In MotifComparison, the default KL threshold has been empirically set to 0.4. 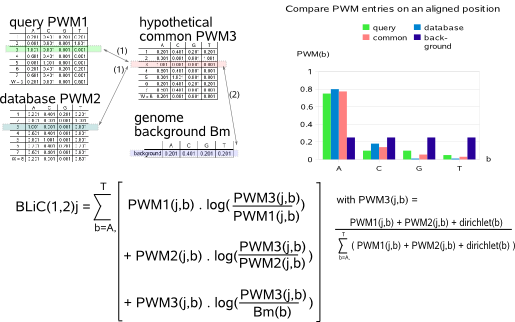 The first term of the BLiC score computes the ratio of the probability
that the query and database motif each are described by a common PWM versus the probability that the two motifs are described by their proper PWM.
The common PWM is unknown and therefore estimated based on the nucleotide counts from the query and database motifs and a Dirichlet prior pseudocount
for each nucleotide A,C,G,T (these counts are described in a file (parameter -p) starting with the symbol '>' followed by underscore separated
values for prior counts on A,C,G,T i.e. >countA_countC_countG_countT). The second term in the BLiC score computes the ratio of the probability that the
query and database motif both correspond to the earlier described common motif versus the probability that the two motifs being compared correspond to the
background. The background is described by the single nucleotide distribution (#snf) in the genome-specific background model (parameter -b). 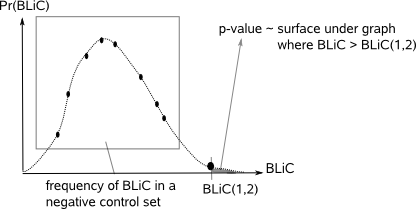
The negative control set for a given query and database motif is obtained by repetitively
(parameter -n times) shuffling the positions in the query and database motif in a random way and computing the BLiC score for the
shuffled motifs after each shuffling. The query and database motif are classified as similar motifs if the p-value of their BLiC score is below
the p-value threshold (parameter -t, default set to 0.001). 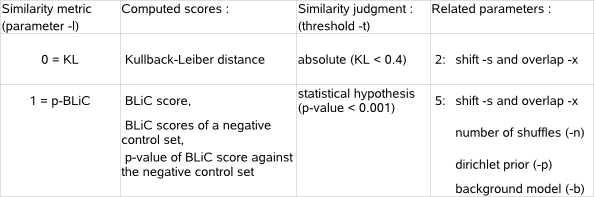 In both cases, whether you choose the KL or the BLiC score, the comparison procedure is repeated for every possible set of query and database motifs present in the two files supplied to MotifComparison. Mark that the admitted set of alignments (determined by parameter -s and -x) between two motifs being compared depends on the length of both motifs and is recomputed for every combination of two motifs that are being compared from the two input files. So at all times, make sure you have reasonable entries for both the shift (-s) and overlap (-x) parameter as this influences the set of evaluated alignments significantly (as demonstrated in Fig.7). 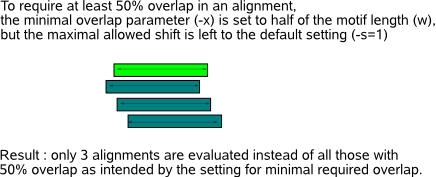 |
Output to the user |
|
The outputfile (parameter -o) of MotifComparison prints the result of comparing each query motif with each database motif on separate lines,
or (choose 'do not print ## lines') only prints the results for query and database motifs that were found similar. |
Evaluation of the output |
- In the KL metric, the same score is computed for equally similar aligned positions regardless if they are informative or not whereas the BLiC score assigns less importance to non-informative compared to informative aligned positions (Fig.8-a). To compensate for overestimating the similarity of two motifs that both have non-informative aligned positions, the default threshold for KL has been set to a stringent value (default -t = 0.4) in MotifComparison. Using a stringent threshold makes the KL metric sensitive to detect true similarities, maybe at the cost of missing some less prominent similarities. The default p-BLiC threshold (-t = 0.001) has a statistical meaning and from our experience, is less stringent than the KL threshold. Repeating MotifComparison with p-BLiC metric thus allows to pick up candidate similarities that were missed by the KL metric, maybe at the expense of recovering some false similarities (i.e. if the dissimilarity of non-informative positions would be underestimated, Fig.8-b). When multiple query motifs were found similar to a given database motif, the magnitude of the BLiC score is most suitable to prioritize the query motifs by their similarity on informative positions to the database motif. 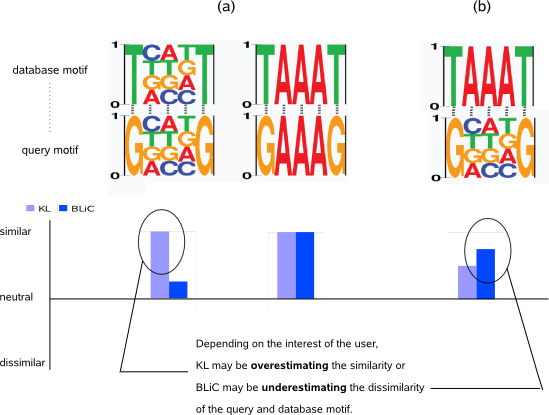
Only for p-BLiC: |
Feedback |
|
Contact us if you have comments, questions or suggestions or simply want to react on the contents of this guideline. Thank you. |