To: Main Page ; To: Application

To: Main Page ; To: Application

Go to evaluation of the output if you need a quick help to optimally run MotifLocator. |
Introduction |
MotifLocator performs a genome-wide screening of DNA sequences for instances of a given motif.
|
MotifLocator Algorithm |
The input for MotifLocator is: 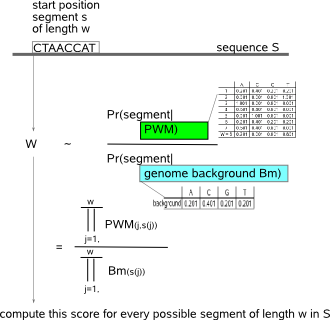
The nominator is computed by multiplying for each position in the segment the frequency with which the nucleotide at this position occurs
according to the frequencies in the PWM of the prior motif. Similarly, the denominator multiplies for each position in the segment the frequency
with which the nucleotide at this position occurs according to the background model. 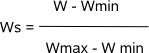
Finally, the segments that have a rescaled score Ws higher than the predefined threshold (parameter -t) are reported as instances for the prior
motif in the selected sequence. The threshold has been empirically set to 0.85 in default mode. The output file reports the computed score of the segments that were selected
as being motif instances of the prior motif. With parameter -a you can choose to display the computed value W of the segment score or the
rescaled value Ws (default) that was used to compare against the score threshold. |
Output to the user |
MotifLocator reports one text output file -o. For each sequence, the selected segments for each prior motif are listed in order of
positional occurrence in the sequence. The format of the file is described in instances format. In short, each line describes |
Evaluation of the output |
- The number of reported instances grows (decreases) exponentially with a decreasing (increasing) threshold. By consequence, the number of
reported instances is sensitive to minor changes in the setting for the threshold (relative to the default setting). The threshold setting must be
between 0 and 1, and the closer to 1 the more stringent the screening (the less instances will be reported). |
Feedback |
|
Contact us if you have comments, questions or suggestions or simply want to react on the contents of this guideline. Thank you. |