To: Main Page ; To: Application

To: Main Page ; To: Application

|
Go to evaluation of the output if you need a quick help to optimally run MotifRanking. |
Introduction |
|
MotifRanking is designed to extract the most likely true motif(s) from a list of multiple motif detection solutions in PWM format. MotifRanking can be used on any list of motifs (PWMs) irrespective of the tool that was used to predict these motifs.
MotifRanking allows prioritizing motifs according to their motif score and their motif detection frequency and computes how many times each motif occurs in the list of predicted motifs (count). |
MotifRanking Algorithm |
|
The input for MotifRanking is a file (parameter -i) containing a list of detected motifs in PWM format
(reported by MotifSampler or any other motif finder). MotifRanking computes two independent metrics to prioritize the detected motifs: the motif score and the motif detection count.  Alternatively, you can also provide your own motif score in the #Score field or MotifRanking can internally compute another score based on the motifs PWM description (Fig.2) : the Consensus score (CS) or the Information Content score (IC). CS is a measure for the conservation of the motif and this score can be used in absolute terms. A perfectly conserved motif has a score equal to 2, while a motif with a uniform nucleotide distribution has a score 0. IC takes not only into account how well the motif is conserved, but also how much the motif differs from the single nucleotide background distribution. To calculate the IC, you need to supply a genome-specific backgroundmodel Bm (format) from which the algorithm will use the zero-order single nucleotide frequencies (#snf). The score is maximal (equals 2) if the motif is well conserved and differs considerably from the background distribution. 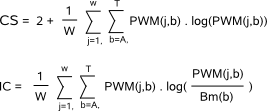 The motif with the highest motif score in the sorted list of detected motifs is compared with each lower sorted motif in this list using a matrix comparison strategy. Two motifs are judged similar if the Kullback-Leiber (KL) distance between their PWMs is below a given similarity threshold (parameter -t). For more details on KL and threshold, we refer to MotifComparison Guidelines. As different alignments between two PWMs may give a different KL distance, all possible alignments (Fig.3) that respect a minimally required overlap (parameter -x) and a maximally allowed shift (parameter -s) are evaluated, 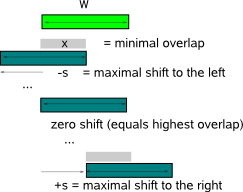 and the best alignment (i.e. with the lowest KL distance) is retained to judge the similarity of the PWMs at hand.
In default mode, -s allows a shift of only 1 nucleotide and -x requires an overlap of at least 6 nucleotides. The reason why two parameters -s and -x are introduced is mainly to handle motif
comparisons of different motif length : allowing for sufficient shifts while conserving a minimal overlap between aligned regions accomodates
the comparison of longer motifs (for which many shifts might be needed to find the optimal alignment) while the minimal overlap is needed to
constrain the allowed shifts for shorter motifs. When all detected motifs have the same length, only one parameter is needed to
restrict the admitted alignments e.g. the minimum overlap constraint (-w) is typically set to half of the (fixed) motif length and the maximal shift (-s)
to an unrestricting high value (e.g. equal to the motif length). 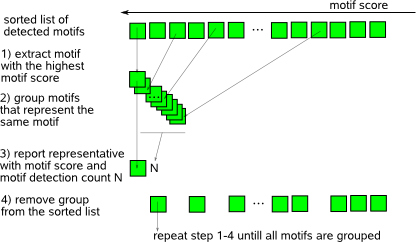 The selection of highest scoring motif, grouping of similar motifs and removal of this group from the list of sorted motifs is repeated untill there are no motifs left that have not been assigned to a particular group of similar motifs. The output of MotifRanking consists of the selected representatives of each group, sorted in order of their motif score together with the size (N) of the group they represent. The return ratio (RR) is a measure for the significance of a detected motif and is obtained by dividing the count N by the total number of motifs that was present in the input file. Mark that the denominator in RR does not necessarily equal the number of initiated motif detection runs (set by MotifSamplers parameter -r) as some runs in MotifSampler may be aborted and report no motif. |
Output to the user |
The retrieved different motifs (default maximal 5, set by parameter -r) are reported in descending motif score order. |
Evaluation of the output |
- At all times, make sure you have reasonable entries for both the shift (-s) and overlap (-x) parameter as this may influence the computation of the motif detection count significantly as demonstrated in Fig.5. 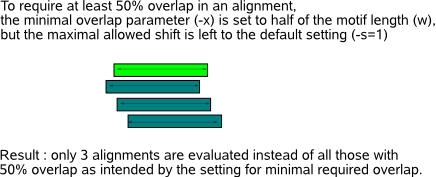 - The fraction of times a same motif was detected by a stochastic motif finder (return ratio RR) is a measure for the significance of a motif,
meaning that motifs with high RR are more statistically relevant and are not likely to have been detected by chance.
>From our experience, highly significant motifs have a return ratio above 50% and motifs with a return ratio below 10% are likely to be spurious.
A high RR also means that the motif to noise signal is strong in the dataset and indicates that the chance is lower that a motif is obscured by untrue instances that do not truly belong to the respective motif. So the higher RR, the better this motif (PWM and instances representation) describes the biologically true motif signal in the dataset. |
Feedback |
|
Contact us if you have comments, questions or suggestions or simply want to react on the contents of this guideline. Thank you. |