To: Main Page ; To: Application

To: Main Page ; To: Application

Go to evaluation of the output if you need a quick help to optimally run CreateConservationPSP. |
Introduction |
|
Related species show similarities in transcriptional regulation, therefore their regulatory sequences are expected to contain the same motif. Adding orthologous sequences to the motif search space is a common pratice to remediate against reporting (suboptimal) solutions with likely no biological functionality (because the predictions are not sufficiently conserved in related species). |
Theoretical background of the algorithm |
|
Three different conservation-based scores were defined by Gordan (Gordan et al., 2009) to compute position specific priors: |
Input for the algorithm |
- a file (parameter -f) containing a set of DNA sequences in FASTA format. CreateConservationPSP is designed to work in combination with MotifSampler. It does so by prioritizing from the MotifSampler DNA input sequences those short DNA segments that have multiple occurrences in other sequences that are expected to be regulated by the same motif. A good candidate for those 'other' sequences are sequences from different species that originated by vertical descent from the same common ancestor as the species of interest for MotifSampler (further called the 'reference species'). Such orthologous sequences in phylogenetically related species can be found in (public) databases. |
CreateConservationPSP Algorithm |
|
The formula in fig.2 describes the computation of the Xc score on each position (p) in a reference sequence (Sr), given a set of M unaligned orthologous sequences (Sm). The use of suffix trees in CreateConservationPSP allows a particularly fast implementation of the string based computation of Xc for any large dataset. 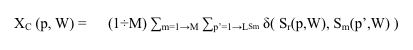
In default mode, Xc scores are computed only for the positions in the reference species. The suffix tree algorithm is repeated for each group of orthologous sequences independently. |
Output to the user |
The algorithm reports one file (parameter -o) with Xc scores in PSP format. The sequence identifier lines (starting with the symbol '>' and/or ">>") in the FASTA file are copied in the PSP file, making the PSP file immediately available for use in MotifSampler. |
Evaluation of the output |
Before using the PSP outputfile as input in MotifSampler, it is recommended to first carefully evaluate the Xc scores in this file. If most Xc scores are high or most Xc scores are low, the PSP is not very informative for use in MotifSampler. Also, if Xc scores are mainly all zero for one sequence and not in others, it may be that the orthologous sequences for the low PSP-scoring sequence are more distant or less well defined compared to the orthologous sequences for the other higher PSP-scoring sequences. To avoid an unfair down-priorization of the low PSP-scoring sequence in MotifSampler, it is better to first delete the PSP information from the PSP file before proceeding with Gibbs sampling based motif detection in MotifSampler. |
References |
- Bailey, T., Boden, M., Whitington, T., & Machanick, P. (2010). The value of position-specific priors in motif discovery using MEME. BMC bioinformatics, 11:179. |
Feedback |
|
Contact us if you have comments, questions or suggestions or simply want to react on the contents of this guideline. Thank you. |