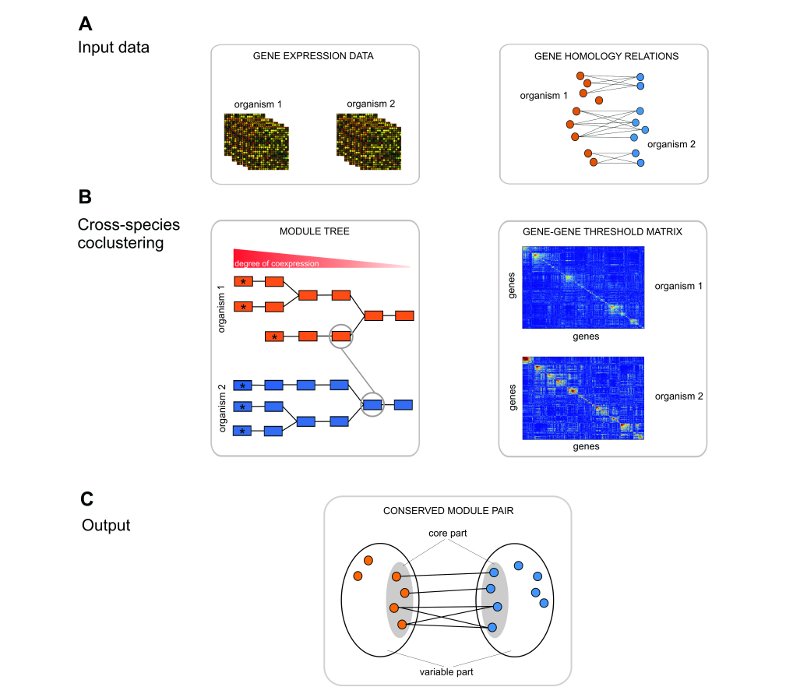
Figure 1. Detection of evolutionary conserved expression modules in a two-species comparison
Installation
1. Download COMODO here
2. Execute the downloaded .exe file, the COMODO v2.0 program will be extracted to the directory in
which the .exe file resides
3. The Matlab Compiler Runtime will be installed, when asked for a .NET installation just press
'continue' (.NET is not needed for COMODO v2.0)
4. Run the unpacked COMODO executable
Usage
COMODO v2.0 requires four input files (see Figure 2):
1. Microarray expression data for species 1 (download sample file here)
2. Microarray expression data for species 2 (same format as file in 1)
3. Microarray expression data for species 3 (same format as file in 1)
4. Homologous gene sets (download sample file here)
The homology definition file (in 4) is a TAB delimited file with the following format:

Five parameters need to be defined in order to run COMODO:
- Initial highest threshold: Defines the maximal coexpression value in the gene-gene threshold matrix. Every coexpression value over this threshold is converted to the given value. It affects the selection of seed module step.
- Least acceptable threshold: Defines the lowest acceptable Pearson correlation coeficient during the seed module extension step.
- Searching step: Defines the stepwise change for relaxing the Paerson correlation coeficient threshold during the seed module extension step.
- Least initial core/total module size: Defines the minimum ratio of the core part over the total module size required for pairing up two seed modules one from each organism.
- Stopping core/total module size: Defines the minimum allowed ratio of the core part over the total module size. It is used as stopping criteria during the seed module extension step.
COMODO v2.0 generates a single output file (download sample file here)
The output consists of three TAB separated columns: MODULE_CORE_PART,ORGANISM1_SPECIFIC_GENES, ORGANISM2_SPECIFIC_GENES and ORGANISM3_SPECIFIC_GENES. The first column also indicates the homology links between genes across organisms in the format (gene1_organism1--gene1_organism2)(gene2_organism1--gene2_organism2) and so on.
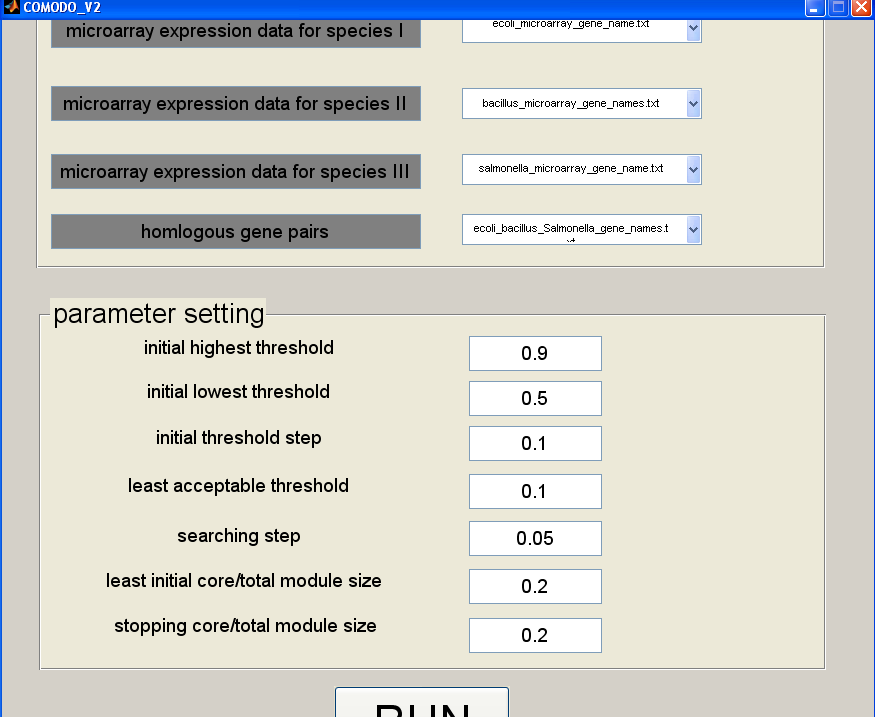
Figure 2. Snapshot of the standalone application
About
This software was developed by the Bioinformatics lab of Prof. K. Marchal at the Centre for Microbial
and Plant Genetics, Faculty of Bioscience Engineering (Katholieke Universiteit Leuven)
Credits
Dr. Peyman Zarrineh
Dr. Carolina Fierro
Dr. Aminael Sanchez-Rodriguez
Zahra Narimani
Nazanin Hosseinkhan
Prof. Ali Masoudi-Nejad
Prof. Kristof Engelen
Prof. Dr. Kathleen Marchal
|
